Understanding and Breaking Scaling Relations in Single-Site Catalysis: Methane to Methanol Conversion by FeIV═O
Abstract
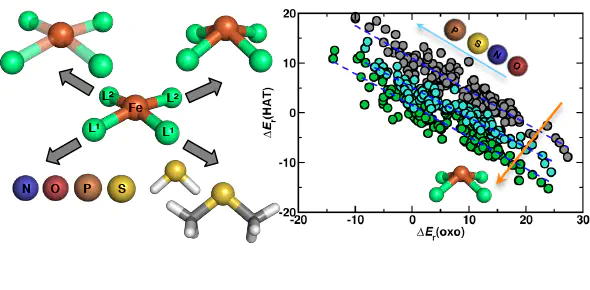
Computational high-throughput screening is an essential tool for catalyst design, limited primarily by the efficiency with which accurate predictions can be made. In bulk heterogeneous catalysis, linear free energy relationships (LFERs) have been extensively developed to relate elementary step activation energies, and thus overall catalytic activity, back to the adsorption energies of key intermediates, dramatically reducing the computational cost of screening. The applicability of these LFERs to single-site catalysts remains unclear, owing to the directional, covalent metal–ligand bonds and the broader chemical space of accessible ligand scaffolds. Through a computational screen of nearly 500 model Fe(II) complexes for CH4 hydroxylation, we observe that (1) tuning ligand field strength yields LFERs by comparably shifting energetics of the metal 3d levels that govern the stability of different intermediates and (2) distortion of the metal coordination geometry breaks these LFERs by increasing the splitting between the dxz/dyz and dz2 metal states that govern reactivity. Thus, in single-site catalysts, low Brønsted–Evans–Polanyi slopes for oxo formation, which would limit peak turnover frequency achievable through ligand field tuning alone, can be overcome through structural distortions achievable in experimentally characterized compounds. Observations from this screen also motivate the placement of strong HB donors in targeted positions as a scaffold-agnostic strategy for further activity improvement. More generally, our findings motivate broader variation of coordination geometries in reactivity studies with single-site catalysts.