Large-scale QM/MM free energy simulations of enzyme catalysis reveal the influence of charge transfer
Abstract
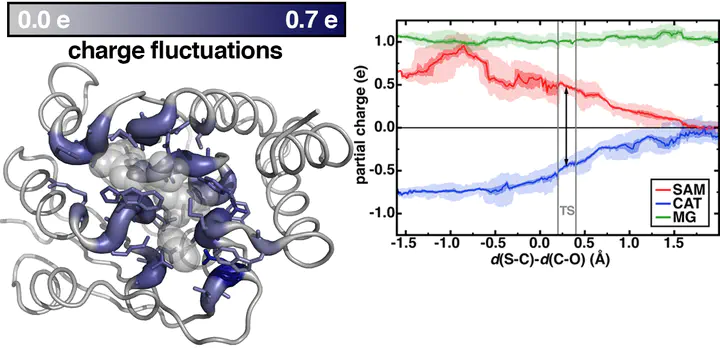
Hybrid quantum mechanical–molecular mechanical (QM/MM) simulations provide key insights into enzyme structure–function relationships. Numerous studies have demonstrated that large QM regions are needed to systematically converge ground state, zero temperature properties with electrostatic embedding QM/MM. However, it is not well known if ab initio QM/MM free energy simulations have this same dependence, in part due to the hundreds of thousands of energy evaluations required for free energy estimations that in turn limit QM region size. Here, we leverage recent advances in electronic structure efficiency and accuracy to carry out range-separated hybrid density functional theory free energy simulations in a representative methyltransferase. By studying 200 ps of ab initio QM/MM dynamics for each of five QM regions from minimal (64 atoms) to one-sixth of the protein (544 atoms), we identify critical differences between large and small QM region QM/MM in charge transfer between substrates and active site residues as well as in geometric structure and dynamics that coincide with differences in predicted free energy barriers. Distinct geometric and electronic structure features in the largest QM region indicate that important aspects of enzymatic rate enhancement in methyltransferases are identified with large-scale electronic structure.