Abstract
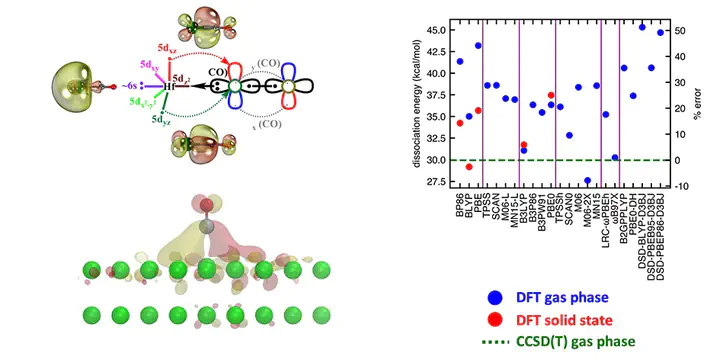
Ab initio multi-reference configuration interaction (MRCI) and coupled cluster singles doubles and perturbative triples [CCSD(T)] levels of theory were used to study ground and excited electronic states of HfCO. We report potential energy curves, dissociation energies (De), excitation energies, harmonic vibrational frequencies, and chemical bonding patterns of HfCO. The 3Σ− ground state of HfCO has an 1σ22σ21π2 electron configuration and a ∼30 kcal mol−1 dissociation energy with respect to its lowest-energy fragments Hf(3F) + CO(X1Σ+). We further evaluated the De of its isovalent HfCX (X = S, Se, Te, Po) series and observed that they increase linearly from the lighter HfCO to the heavier HfCPo with the dipole moment of the CX ligand. The same linear relationship was observed for TiCX and ZrCX. We utilized the CCSD(T) benchmark values of De, excitation energy, and ionization energy (IE) values to evaluate density functional theory (DFT) errors with 23 exchange–correlation functionals spanning GGA, meta-GGA, global GGA hybrid, meta-GGA hybrid, range-separated hybrid, and double-hybrid functional families. The global GGA hybrid B3LYP and range-separated hybrid ωB97X performed well at representing the ground state properties of HfCO (i.e., De and IE). Finally, we extended our DFT analysis to the interaction of a CO molecule with a Hf surface and observed that the surface chemisorption energy and the gas-phase molecular dissociation energy are very similar for some DFAs but not others, suggesting moderate transferability of the benchmarks on these molecules to the solid state.