Molecular DFT+U: A Transferable, Low-Cost Approach to Eliminate Delocalization Error
Abstract
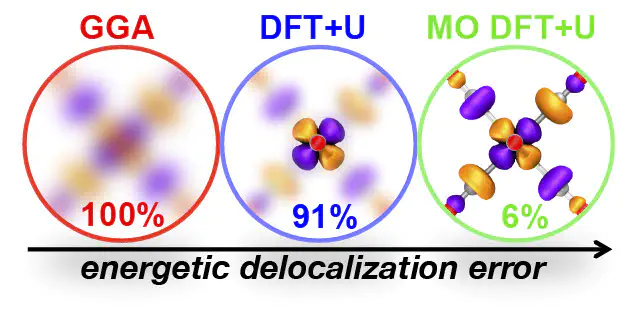
While density functional theory (DFT) is widely applied for its combination of cost and accuracy, corrections (e.g., DFT+U) that improve it are often needed to tackle correlated transition-metal chemistry. In principle, the functional form of DFT+U, consisting of a set of localized atomic orbitals (AOs) and a quadratic energy penalty for deviation from integer occupations of those AOs, enables the recovery of the exact conditions of piecewise linearity and the derivative discontinuity. Nevertheless, for practical transition-metal complexes, where both atomic states and ligand orbitals participate in bonding, standard DFT+U can fail to eliminate delocalization error (DE). Here, we show that by introducing an alternative valence-state (i.e., molecular orbital or MO) basis to the DFT+U approach, we recover exact conditions in cases for which standard DFT+U corrections have no error-reducing effect. This MO-based DFT+U also eliminates DE where standard AO-based DFT+U is already successful. We demonstrate the transferability of our approach on representative transition-metal complexes with a range of ligand field strengths, electron configurations (i.e., from Sc to Zn), and spin states.