Learning from Failure: Predicting Electronic Structure Calculation Outcomes with Machine Learning Models
Abstract
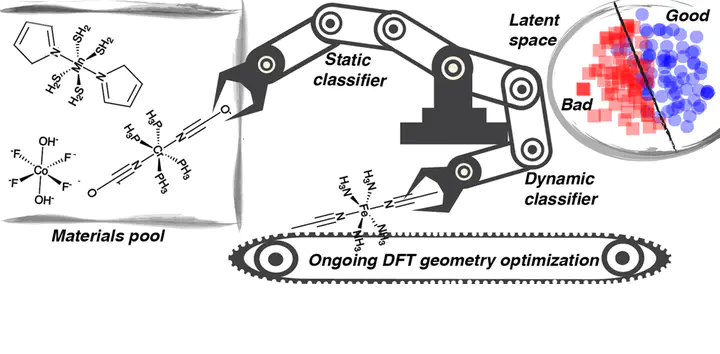
High-throughput computational screening for chemical discovery mandates the automated and unsupervised simulation of thousands of new molecules and materials. In challenging materials spaces, such as open shell transition metal chemistry, characterization requires time-consuming first-principles simulation that often necessitates human intervention. These calculations can frequently lead to a null result, e.g., the calculation does not converge or the molecule does not stay intact during a geometry optimization. To overcome this challenge toward realizing fully automated chemical discovery in transition metal chemistry, we have developed the first machine learning models that predict the likelihood of successful simulation outcomes. We train support vector machine and artificial neural network classifiers to predict simulation outcomes (i.e., geometry optimization result and degree of ⟨S2⟩ deviation) for a chosen electronic structure method based on chemical composition. For these static models, we achieve an area under the curve of at least 0.95, minimizing computational time spent on nonproductive simulations and therefore enabling efficient chemical space exploration. We introduce a metric of model uncertainty based on the distribution of points in the latent space to systematically improve model prediction confidence. In a complementary approach, we train a convolutional neural network classification model on simulation output electronic and geometric structure time series data. This dynamic model generalizes more readily than the static classifier by becoming more predictive as input simulation length increases. Finally, we describe approaches for using these models to enable autonomous job control in transition metal complex discovery.