Machine learning to tame divergent density functional approximations a new path to consensus materials design principles
Abstract
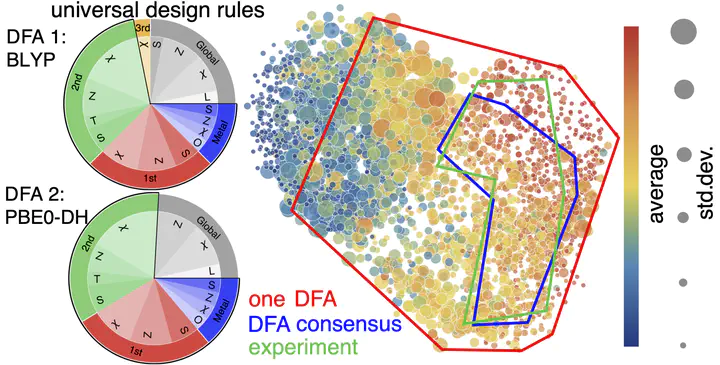
Virtual high-throughput screening (VHTS) with density functional theory (DFT) and machine-learning (ML)-acceleration is essential in rapid materials discovery. By necessity, efficient DFT-based workflows are carried out with a single density functional approximation (DFA). Nevertheless, properties evaluated with different DFAs can be expected to disagree for cases with challenging electronic structure (e.g., open-shell transition-metal complexes, TMCs) for which rapid screening is most needed and accurate benchmarks are often unavailable. To quantify the effect of DFA bias, we introduce an approach to rapidly obtain property predictions from 23 representative DFAs spanning multiple families, “rungs” (e.g., semi-local to double hybrid) and basis sets on over 2000 TMCs. Although computed property values (e.g., spin state splitting and frontier orbital gap) differ by DFA, high linear correlations persist across all DFAs. We train independent ML models for each DFA and observe convergent trends in feature importance, providing DFA-invariant, universal design rules. We devise a strategy to train artificial neural network (ANN) models informed by all 23 DFAs and use them to predict properties (e.g., spin-splitting energy) of over 187k TMCs. By requiring consensus of the ANN-predicted DFA properties, we improve correspondence of computational lead compounds with literature-mined, experimental compounds over the typically employed single-DFA approach.