Machine learning models predict calculation outcomes with the transferability necessary for computational catalysis
Abstract
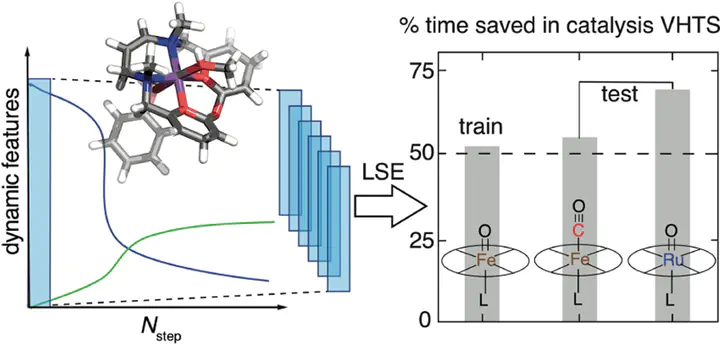
Virtual high-throughput screening (VHTS) and machine learning (ML) have greatly accelerated the design of single-site transition-metal catalysts. VHTS of catalysts, however, is often accompanied with high calculation failure rate and wasted computational resources due to the difficulty of simultaneously converging all mechanistically relevant reactive intermediates to expected geometries and electronic states. We demonstrate a dynamic classifier approach, i.e., a convolutional neural network that monitors geometry optimizations on the fly, and exploit its good performance and transferability on identifying geometry optimization failures for catalyst design. We show that the dynamic classifier performs well on all reactive intermediates in the representative catalytic cycle of the radical rebound mechanism for the conversion of methane to methanol despite being trained on only one reactive intermediate. The dynamic classifier also generalizes to chemically distinct intermediates and metal centers absent from the training data without loss of accuracy or model confidence. We rationalize this superior model transferability as arising from the use of on-the-fly electronic structure and geometric information generated from on-the-fly density functional theory calculations and the convolutional layer in the dynamic classifier. When used in combination with uncertainty quantification, the dynamic classifier saves more than half of the computational resources that would have been wasted on unsuccessful calculations for all reactive intermediates being considered.