Putting Density Functional Theory to the Test in Machine-Learning-Accelerated Materials Discovery
Abstract
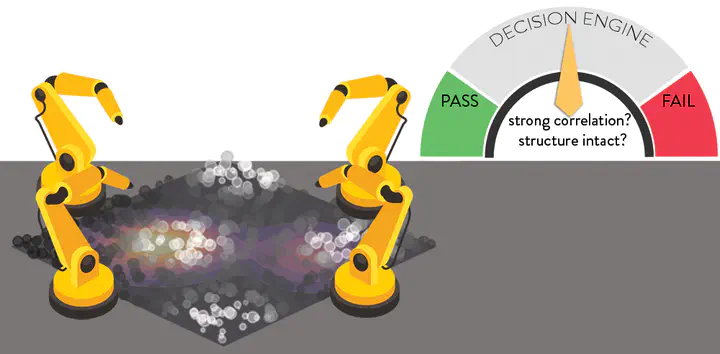
Accelerated discovery with machine learning (ML) has begun to provide the advances in efficiency needed to overcome the combinatorial challenge of computational materials design. Nevertheless, ML-accelerated discovery both inherits the biases of training data derived from density functional theory (DFT) and leads to many attempted calculations that are doomed to fail. Many compelling functional materials and catalytic processes involve strained chemical bonds, open-shell radicals and diradicals, or metal–organic bonds to open-shell transition-metal centers. Although promising targets, these materials present unique challenges for electronic structure methods and combinatorial challenges for their discovery. In this Perspective, we describe the advances needed in accuracy, efficiency, and approach beyond what is typical in conventional DFT-based ML workflows. These challenges have begun to be addressed through ML models trained to predict the results of multiple methods or the differences between them, enabling quantitative sensitivity analysis. For DFT to be trusted for a given data point in a high-throughput screen, it must pass a series of tests. ML models that predict the likelihood of calculation success and detect the presence of strong correlation will enable rapid diagnoses and adaptation strategies. These “decision engines” represent the first steps toward autonomous workflows that avoid the need for expert determination of the robustness of DFT-based materials discoveries.