A Transferable Recommender Approach for Selecting the Best Density Functional Approximations in Chemical Discovery
Abstract
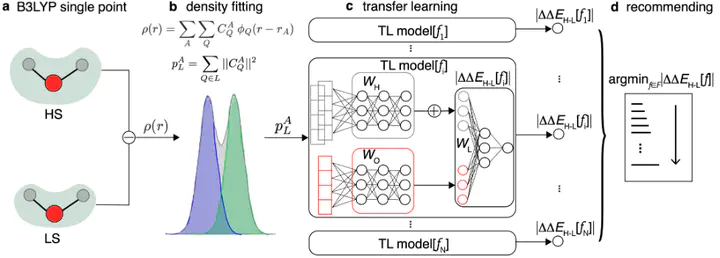
Approximate density functional theory (DFT) has become indispensable owing to its cost-accuracy trade-off in comparison to more computationally demanding but accurate correlated wavefunction theory. To date, however, no single density functional approximation (DFA) with universal accuracy has been identified, leading to uncertainty in the quality of data generated from DFT. With electron density fitting and transfer learning, we build a DFA recommender that selects the DFA with the lowest expected error with respect to gold standard but cost-prohibitive coupled cluster theory in a system-specific manner. We demonstrate this recommender approach on vertical spin-splitting energy evaluation for challenging transition metal complexes. Our recommender predicts top-performing DFAs and yields excellent accuracy (ca. 2 kcal/mol) for chemical discovery, outperforming both individual transfer learning models and the single best functional in a set of 48 DFAs. We demonstrate the transferability of the DFA recommender to experimentally synthesized compounds with distinct chemistry.