Unifying Exchange Sensitivity in Transition-Metal Spin-State Ordering and Catalysis through Bond Valence Metrics
Abstract
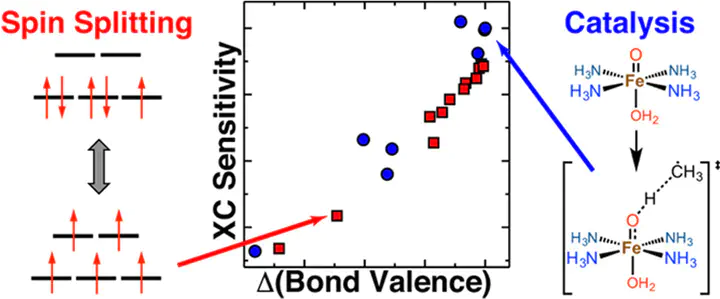
Accurate predictions of spin-state ordering, reaction energetics, and barrier heights are critical for the computational discovery of open-shell transition-metal (TM) catalysts. Semilocal approximations in density functional theory, such as the generalized gradient approximation (GGA), suffer from delocalization error that causes them to overstabilize strongly bonded states. Descriptions of energetics and bonding are often improved by introducing a fraction of exact exchange (e.g., erroneous low-spin GGA ground states are instead correctly predicted as high-spin with a hybrid functional). The degree of spin-splitting sensitivity to exchange can be understood based on the chemical composition of the complex, but the effect of exchange on reaction energetics within a single spin state is less well-established. Across a number of model iron complexes, we observe strong exchange sensitivities of reaction barriers and energies that are of the same magnitude as those for spin splitting energies. We rationalize trends in both reaction and spin energetics by introducing a measure of delocalization, the bond valence of the metal–ligand bonds in each complex. The bond valence thus represents a simple-to-compute property that unifies understanding of exchange sensitivity for catalytic properties and spin-state ordering in TM complexes. Close agreement of the resulting per-metal–organic-bond sensitivity estimates, together with failure of alternative descriptors demonstrates the utility of the bond valence as a robust descriptor of how differences in metal–ligand delocalization produce differing relative energetics with exchange tuning. Our unified description explains the overall effect of exact exchange tuning on the paradigmatic two-state FeO+/CH4 reaction that combines challenges of spin-state and reactivity predictions. This new descriptor-sensitivity relationship provides a path to quantifying how predictions in transition-metal complex screening are sensitive to the method used.