Accelerating Chemical Discovery with Machine Learning: Simulated Evolution of Spin Crossover Complexes with an Artificial Neural Network
Abstract
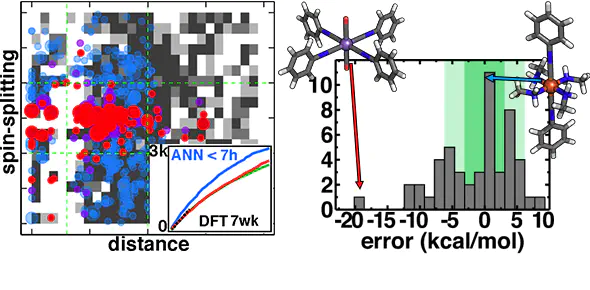
Machine learning (ML) has emerged as a powerful complement to simulation for materials discovery by reducing time for evaluation of energies and properties at accuracy competitive with first-principles methods. We use genetic algorithm (GA) optimization to discover unconventional spin-crossover complexes in combination with efficient scoring from an artificial neural network (ANN) that predicts spin-state splitting of inorganic complexes. We explore a compound space of over 5600 candidate materials derived from eight metal/oxidation state combinations and a 32-ligand pool. We introduce a strategy for error-aware ML-driven discovery by limiting how far the GA travels away from the nearest ANN training points while maximizing property (i.e., spin-splitting) fitness, leading to discovery of 80% of the leads from full chemical space enumeration. Over a 51-complex subset, average unsigned errors (4.5 kcal/mol) are close to the ANN’s baseline 3 kcal/mol error. By obtaining leads from the trained ANN within seconds rather than days from a DFT-driven GA, this strategy demonstrates the power of ML for accelerating inorganic material discovery.