Leveraging Cheminformatics Strategies for Inorganic Discovery: Application to Redox Potential Design
Abstract
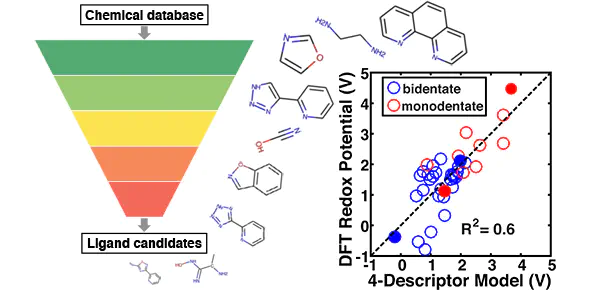
Virtual high throughput screening, typically driven by first-principles, density functional theory calculations, has emerged as a powerful tool for the discovery of new materials. Although the computational materials science community has benefited from open source tools for the rapid structure generation, calculation, and analysis of crystalline inorganic materials, software and strategies to address the unique challenges of inorganic complex discovery have not been as widely available. We present a unified view of our recent developments in the open source molSimplify code for inorganic discovery. Building on our previous efforts in the automated generation of highly accurate inorganic molecular structures, first-principles simulation, and property analysis to accelerate high-throughput screening, we have recently incorporated a neural network that both improves structure generation and predicts electronic properties prior to first-principles calculation. We also provide an overview of how multimillion molecule organic libraries can be leveraged for inorganic discovery alongside cheminformatics concepts of molecular diversity in order to efficiently traverse chemical space. We demonstrate all of these tools on the discovery of design rules for octahedral Fe(II/III) redox couples with nitrogen ligands. Over a search of only approximately 40 new molecules, we obtain redox potentials relative to the Fc/Fc+ couple ranging from −1 to 4.5 V in aqueous solution. Our new automated correlation analysis reveals heteroatom identity and the degree of structural branching to be key ligand descriptors in determining redox potential. This inorganic discovery toolkit provides a promising approach to advancing transition metal complex design.