A quantitative uncertainty metric controls error in neural network-driven chemical discovery
Abstract
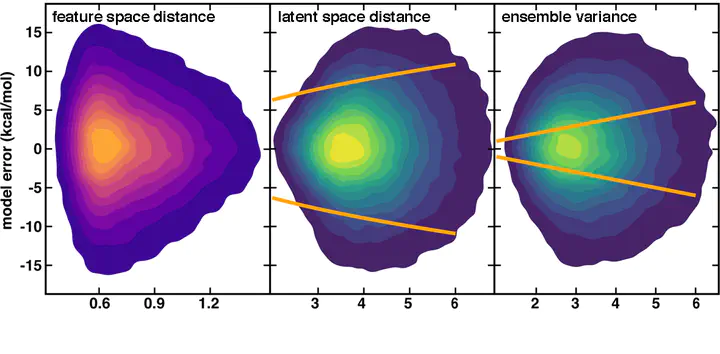
Machine learning (ML) models, such as artificial neural networks, have emerged as a complement to high-throughput screening, enabling characterization of new compounds in seconds instead of hours. The promise of ML models to enable large-scale chemical space exploration can only be realized if it is straightforward to identify when molecules and materials are outside the model’s domain of applicability. Established uncertainty metrics for neural network models are either costly to obtain (e.g., ensemble models) or rely on feature engineering (e.g., feature space distances), and each has limitations in estimating prediction errors for chemical space exploration. We introduce the distance to available data in the latent space of a neural network ML model as a low-cost, quantitative uncertainty metric that works for both inorganic and organic chemistry. The calibrated performance of this approach exceeds widely used uncertainty metrics and is readily applied to models of increasing complexity at no additional cost. Tightening latent distance cutoffs systematically drives down predicted model errors below training errors, thus enabling predictive error control in chemical discovery or identification of useful data points for active learning.