Abstract
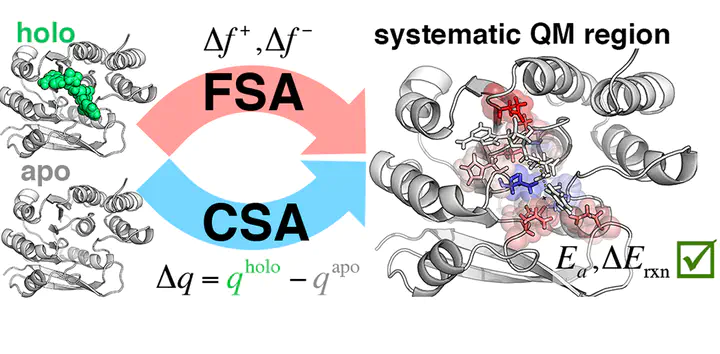
Hybrid quantum mechanical-molecular mechanical (QM/MM) simulations are widely used in enzyme simulation. Over ten convergence studies of QM/MM methods have revealed over the past several years that key energetic and structural properties approach asymptotic limits with only very large (ca. 500–1000 atom) QM regions. This slow convergence has been observed to be due in part to significant charge transfer between the core active site and the surrounding protein environment, which cannot be addressed by improvement of MM force fields or the embedding method employed within QM/MM. Given this slow convergence, it becomes essential to identify strategies for the most atom-economical determination of optimal QM regions and to gain insight into the crucial interactions captured only in large QM regions. Here, we extend and develop two methods for quantitative determination of QM regions. First, in the charge shift analysis (CSA) method, we probe the reorganization of electron density when core active site residues are removed completely, as determined by large-QM region QM/MM calculations. Second, we introduce the highly parallelizable Fukui shift analysis (FSA), which identifies how core/substrate frontier states are altered by the presence of an additional QM residue in smaller initial QM regions. We demonstrate that the FSA and CSA approaches are complementary and consistent on three test case enzymes: catechol O-methyltransferase, cytochrome P450cam, and hen eggwhite lysozyme. We also introduce validation strategies and test the sensitivities of the two methods to geometric structure, basis set size, and electronic structure methodology. Both methods represent promising approaches for the systematic, unbiased determination of quantum mechanical effects in enzymes and large systems that necessitate multiscale modeling.