Abstract
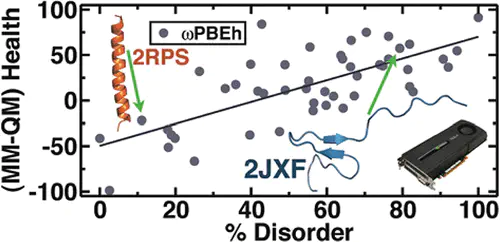
Structural properties of over 55 small proteins have been determined using both density-based and wave-function-based electronic structure methods in order to assess the ability of ab initio “force fields” to retain the properties described by experimental structures measured with crystallography or nuclear magnetic resonance. The efficiency of the GPU-based quantum chemistry algorithms implemented in our TeraChem program enables us to carry out systematic optimization of ab initio protein structures, which we compare against experimental and molecular mechanics force field references. We show that the quality of the ab initio optimized structures, as judged by conventional protein health metrics, increases with increasing basis set size. On the other hand, there is little evidence for a significant improvement of predicted structures using density functional theory as compared to Hartree–Fock methods. Although occasional pathologies of minimal basis sets are observed, these are easily alleviated with even the smallest double-ζ basis sets.