Density Functional Theory in Transition-Metal Chemistry: A Self-Consistent Hubbard U Approach
Abstract
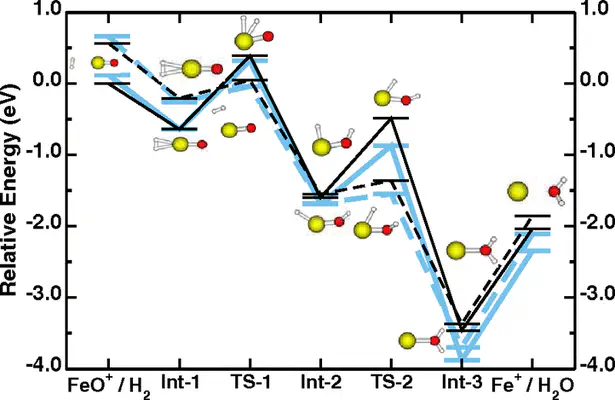
Transition-metal centers are the active sites for a broad variety of biological and inorganic chemical reactions. Notwithstanding this central importance, density-functional theory calculations based on generalized-gradient approximations often fail to describe energetics, multiplet structures, reaction barriers, and geometries around the active sites. We suggest here an alternative approach, derived from the Hubbard U correction to solid-state problems, that provides an excellent agreement with correlated-electron quantum chemistry calculations in test cases that range from the ground state of Fe2 and Fe−2 to the addition elimination of molecular hydrogen on FeO+. The Hubbard U is determined with a novel self-consistent procedure based on a linear-response approach.