Making machine learning a useful tool in the accelerated discovery of transition metal complexes
Abstract
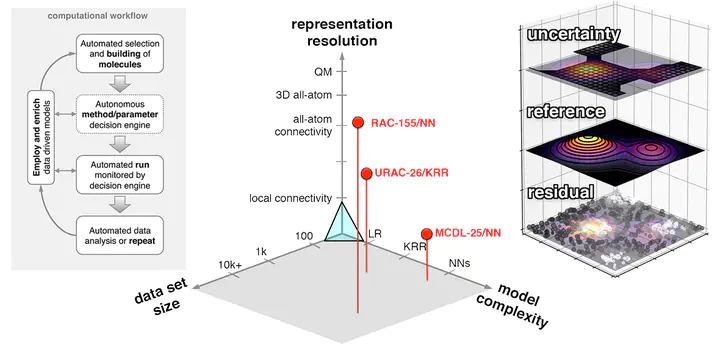
As machine learning (ML) has matured, it has opened a new frontier in theoretical and computational chemistry by offering the promise of simultaneous paradigm shifts in accuracy and efficiency. Nowhere is this advance more needed, but also more challenging to achieve, than in the discovery of open-shell transition metal complexes. Here, localized d or f electrons exhibit variable bonding that is challenging to capture even with the most computationally demanding methods. Thus, despite great promise, clear obstacles remain in constructing ML models that can supplement or even replace explicit electronic structure calculations. In this article, I outline the recent advances in building ML models in transition metal chemistry, including the ability to approach sub-kcal/mol accuracy on a range of properties with tailored representations, to discover and enumerate complexes in large chemical spaces, and to reveal opportunities for design through analysis of feature importance. I discuss unique considerations that have been essential to enabling ML in open-shell transition metal chemistry, including (a) the relationship of data set size/diversity, model complexity, and representation choice, (b) the importance of quantitative assessments of both theory and model domain of applicability, and (c) the need to enable autonomous generation of reliable, large data sets both for ML model training and in active learning or discovery contexts. Finally, I summarize the next steps toward making ML a mainstream tool in the accelerated discovery of transition metal complexes. This article is categorized under: Electronic Structure Theory textgreater Density Functional Theory Software textgreater Molecular Modeling Computer and Information Science textgreater Chemoinformatics