Abstract
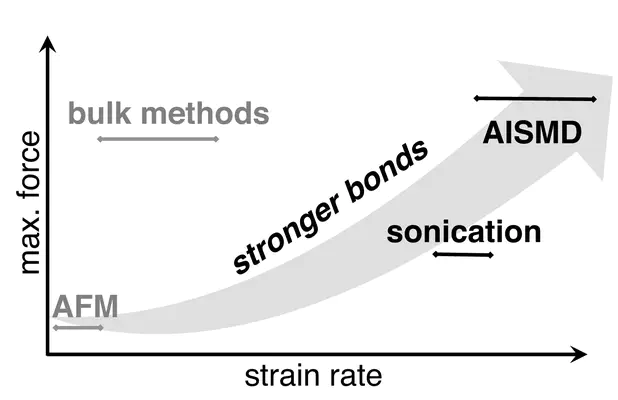
This chapter first defines fundamental concepts governing chemical transformation on a potential energy surface (PES). Then, it introduces theoretical and computational methodology that incorporates the effect of mechanical force in order to determine its effect on chemical transformation rates and pathways. This theoretical and computational background is followed by a discussion of the basics of mechanochemical experiments, including both approaches and the nature of molecules studied. This background then enables the readers to review some fundamental, representative cases of how computation has provided insight into mechanochemical bond cleavage. The chapter further provides a summary of practical challenges for the novice wishing to carry out mechanochemical simulation. While relevant for all fields of simulation, a particularly pertinent concern in the first-principles modeling of mechanochemical bond cleavage is the balance of computational cost and accuracy. The chapter concludes with an overview of the current status of computational mechanochemistry and related approaches.