A self-consistent Hubbard U density-functional theory approach to the addition-elimination reactions of hydrocarbons on bare FeO+
Abstract
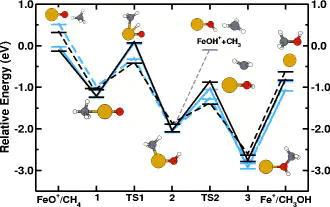
We present a detailed analysis of the addition-elimination reaction pathways for the gas-phase conversion of molecular hydrogen and methane on FeO+ to water and methanol, respectively, using first-principles calculations. These two reactions represent paradigmatic, challenging test cases for electronic structure approaches to transition-metal catalysis. We compare here density-functional approaches against state-of-the-art coupled-cluster and multireference quantum chemistry approaches. The quantum chemical approaches are found to be in close agreement between themselves as well as with the available experimental evidence. For the density-functional calculations, we employ a recently introduced ab initio, self-consistent Hubbard-like correction, coupled here with a generalized-gradient approximation (GGA) for the exchange-correlation functional. We find that our formulation provides a remarkable improvement in the description of the electronic structure, hybridization, and multiplet splittings for all calculated stationary points along these reaction pathways. The Hubbard term, which is not a fitting parameter and, in principle, can augment any exchange-correlation functional, brings the density-functional theory results in close agreement with the reference calculations. In particular, thermochemical errors as large as 1.4 eV in the exit channels with the GGA functional are reduced by an order of magnitude, to less than 0.1 eV on average; additionally, close agreement with the correlated-electron reference calculations and experiments are achieved for intermediate spin splittings and structures, reaction exothermicity, and spin crossovers. The role that the Hubbard U term plays in improving both quantitative and qualitative descriptions of transition-metal chemistry is examined, and its strengths as well as possible weaknesses are discussed in detail.