Abstract
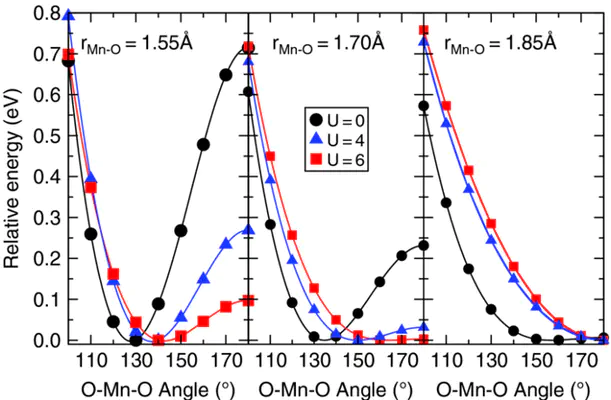
Triatomic transition-metal oxides in the “inserted dioxide” (O–M–O) structure represent one of the simplest examples of systems that undergo qualitative geometrical changes via subtle electronic-structure modulation. We consider here three transition-metal dioxide molecules (MO2 where M = Mn, Fe, or Co), for which the equilibrium structural (e.g., bent or linear geometry) and electronic (e.g., spin or symmetry) properties have been challenging to assign both theoretically and experimentally. Augmenting a standard density-functional theory (DFT) approach with a Hubbard term (DFT+U) occasionally overlocalizes the 3d manifold, leading to an incorrect bond elongation and, in turn, poor equilibrium geometries for MO2 molecules, while preserving good spin-state splittings. Proper description of both geometry and energetics for these molecules is recovered; however, through either calculating DFT+U relaxations at fixed M–O bond lengths or by inclusion of an intersite interaction term V that favors M(3d)–O(2p) interactions. In this latter case, both U and V are calculated fully from first-principles and are not fitting parameters. Finally, we identify an approach that more accurately determines the Hubbard U over a coordinate in which the covalent character of bonding varies.