Bridging the Homogeneous-Heterogeneous Divide: Modeling Spin for Reactivity in Single Atom Catalysis
Abstract
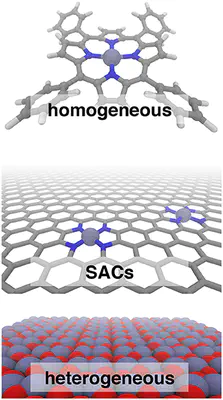
Single atom catalysts (SACs) are emergent catalytic materials that have the promise of merging the scalability of heterogeneous catalysts with the high activity and atom economy of homogeneous catalysts. Computational, first-principles modeling can provide essential insight into SAC mechanism and active site configuration, where the sub-nm-scale environment can challenge even the highest-resolution experimental spectroscopic techniques. Nevertheless, the very properties that make SACs attractive in catalysis, such as localized d electrons of the isolated transition metal center, make them challenging to study with conventional computational modeling using density functional theory (DFT). For example, Fe/N-doped graphitic SACs have exhibited spin-state dependent reactivity that remains poorly understood. However, spin-state ordering in DFT is very sensitive to the nature of the functional approximation chosen. In this work, we develop accurate benchmarks from correlated wavefunction theory (WFT) for relevant octahedral complexes. We use those benchmarks to evaluate optimal DFT functional choice for predicting spin state ordering in small octahedral complexes as well as models of pyridinic and pyrrolic nitrogen environments expected in larger SACs. Using these guidelines, we determine Fe/N-doped graphene SAC model properties and reactivity as well as their sensitivities to DFT functional choice. Finally, we conclude with broad recommendations for computational modeling of open-shell transition metal single-atom catalysts.