Impact of Approximate DFT Density Delocalization Error on Potential Energy Surfaces in Transition Metal Chemistry
Abstract
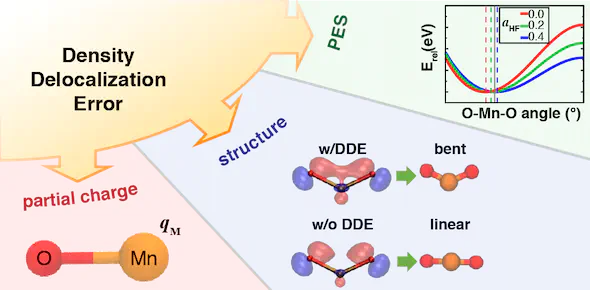
For approximate density functional theory (DFT) to be useful in catalytic applications of transition metal complexes, modeling strategies must simultaneously address electronic, geometric, and energetic properties of the relevant species. We show that for representative transition metal triatomics (MO2, where M = Cr, Mn, Fe, Co, or Ni) and related diatomics the incorporation of Hartree–Fock (HF) exchange in most cases improves the properties of the Born–Oppenheimer potential energy surface (PES) with respect to accurate experimental or CCSD(T) references. We rationalize this observation by noting reduced delocalization obtained with hybrid functionals (20–40% HF exchange), as evidenced by reduced hybridization of non-bonding orbitals and increases in metal partial charges. Although we show that the optimal exchange fraction is both property and system specific, incorporating HF exchange synergistically improves properties of density, structure, and energetics within a single PES characterized by moderately covalent bonding. The same improvement is not observed in the ordering of MO2 spin states, as good agreement of semi-local DFT spin state ordering is worsened by over-stabilization of higher spin states when HF exchange is added. More work is needed to understand minimal functional forms capable of improving multiple properties with respect to semi-local DFT descriptions of transition metal chemistry.