Large-scale Screening Reveals Geometric Structure Matters More than Electronic Structure in Bioinspired Catalyst Design of Formate Dehydrogenase Mimics
Abstract
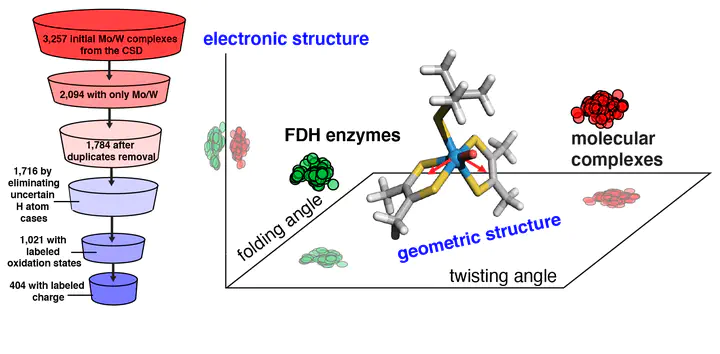
The design of inorganic molecular complexes for the reversible conversion of formate into CO2 inspired by formate dehydrogenase (FDH) enzymes is challenged by a lack of understanding of how to mimic the enzyme action. Here, we carry out a large-scale, high-throughput screening study on all mononuclear Mo/W complexes currently deposited in Cambridge Structural Database (CSD) that resemble the coordination environment of the molybdopterin cofactors in FDH. Using density functional theory, we systematically investigate the individual effects of metal identity, ligand identity, oxidation state, and coordination number on structural, electronic, and catalytic (i.e., H atom binding) properties. We compare our results on molecular complexes with large quantum mechanical cluster calculations on a representative FDH enzyme to understand the influence of the enzyme environment. These comparisons reveal that the enzyme environment primarily influences the metal-local geometry, and these structural variations can improve catalysis. Through a series of computational substitutions on molecular complexes of terminal chalcogen atoms and metal centers, we extend beyond CSD structures to identify the limits of varied chalcogen and metal identity. Through this analysis, we demonstrate that the enzyme primarily affects the geometric properties of the metal center, and terminal chalcogen moieties primarily influence local electronic properties.