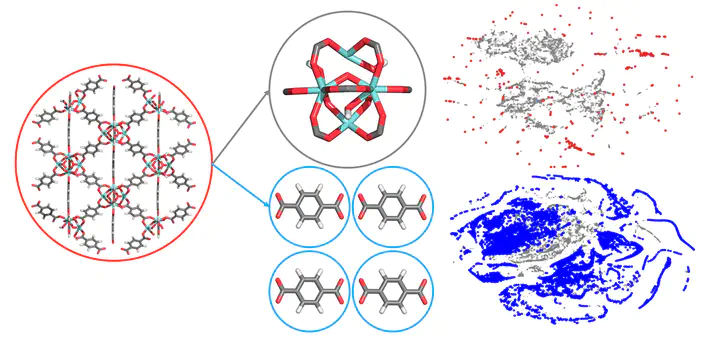
Abstract
Millions of distinct metal-organic frameworks (MOFs) can be made by combining metal nodes and organic linkers. At present, over 90,000 MOFs have been synthesized and over 500,000 predicted. This raises the question whether a new experimental or predicted structure adds new information. For MOF chemists, the chemical design space is a combination of pore geometry, metal nodes, organic linkers, and functional groups, but at present we do not have a formalism to quantify optimal coverage of chemical design space. In this work, we develop a machine learning method to quantify similarities of MOFs to analyse their chemical diversity. This diversity analysis identifies biases in the databases, and we show that such bias can lead to incorrect conclusions. The developed formalism in this study provides a simple and practical guideline to see whether new structures will have the potential for new insights, or constitute a relatively small variation of existing structures.