Why Conventional Design Rules for C–H Activation Fail for Open-Shell Transition-Metal Catalysts
Abstract
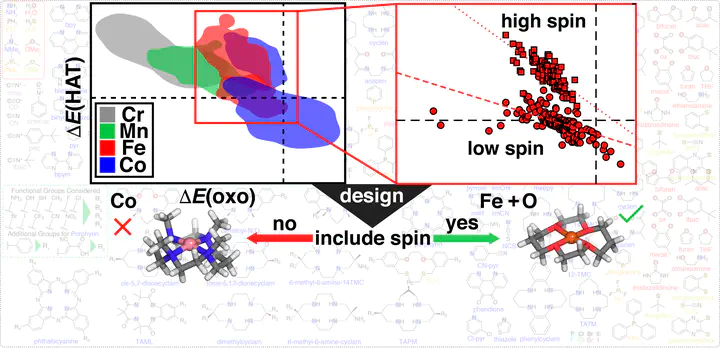
The design of selective and active C–H activation catalysts for direct methane-to-methanol conversion is challenging. Bioinspired complexes that form high-valent metal–oxo intermediates capable of hydrogen abstraction and rebound hydroxylation are promising candidates. This promise has made them a target for computational high-throughput screening, typically simplified through the use of linear free energy relationships (LFERs). However, their mid-row transition-metal centers have numerous accessible spin and oxidation states that increase the combinatorial scale of design efforts. Here, we carry out a computational design screen of over 2500 mid-row 3d transition-metal complexes with four metals in numerous spin and oxidation states. We demonstrate the importance of spin/oxidation state in dictating design principles, limiting the generalization of strategies derived for widely studied high-spin Fe(II) catalysts to other metals or spin/oxidation states. Combined assessment of the effect of ligand-field tuning on reaction step energetics and on the identity of the ground state allows us to propose refined design strategies for spin-allowed methane-to-methanol catalysis. We observe weak coupling of energetics and design principles between reaction steps (e.g., oxo formation vs methanol release), meaning that LFERs do not generalize across our larger catalyst set. To rationalize relative reactivity in known catalysts, we instead compute independent reaction energies and propose strategies for further improvements in catalyst design.