Evaluating Unexpectedly Short Non-covalent Distances in X-ray Crystal Structures of Proteins with Electronic Structure Analysis
Abstract
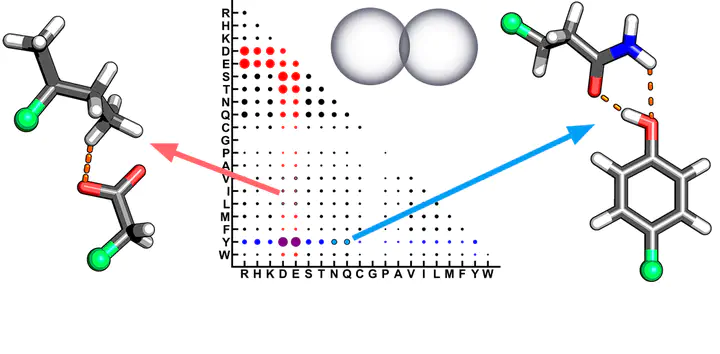
We investigate unexpectedly short non-covalent distances (<85% of the sum of van der Waals radii) in X-ray crystal structures of proteins. We curate over 11 000 high-quality protein crystal structures and an ultra-high-resolution (1.2 Å or better) subset containing >900 structures. Although our non-covalent distance criterion excludes standard hydrogen bonds known to be essential in protein stability, we observe over 75 000 close contacts (CCs) in the curated protein structures. Analysis of the frequency of amino acids participating in these interactions demonstrates some expected trends (i.e., enrichment of charged Lys, Arg, Asp, and Glu) but also reveals unexpected enhancement of Tyr in such interactions. Nearly all amino acids are observed to form at least one CC with all other amino acids, and most interactions are preserved in the much smaller ultra-high-resolution subset. We quantum-mechanically characterize the interaction energetics of a subset of >5000 CCs with symmetry-adapted perturbation theory to enable decomposition of interactions. We observe the majority of CCs to be favorable. The shortest favorable non-covalent distances are under 2.2 Å and are very repulsive when characterized with classical force fields. This analysis reveals stabilization by a combination of electrostatic and charge-transfer effects between hydrophobic (i.e., Val, Ile, Leu) amino acids and charged Asp or Glu. We also observe a unique hydrogen-bonding configuration between Tyr and Asn/Gln involving both residues acting simultaneously as hydrogen bond donors and acceptors. This work confirms the importance of first-principles simulation in explaining unexpected geometries in protein crystal structures.