Mapping the Origins of Surface- and Chemistry-Dependent Doping Trends in III-V Quantum Dots with Density Functional Theory
Abstract
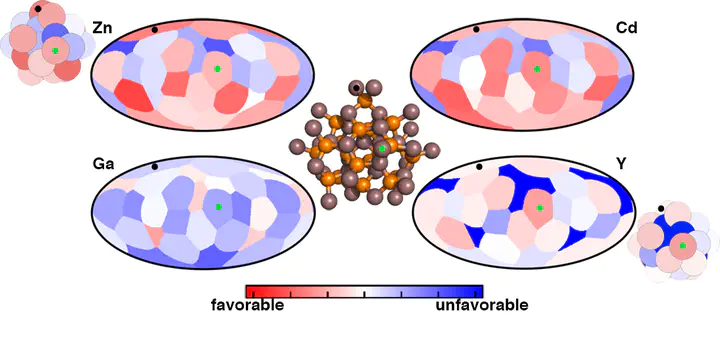
Modifying the optoelectronic properties of nanostructured materials through the introduction of dopant atoms has attracted intense interest, but the approaches employed are often trial and error, preventing rational design. We demonstrate the power of large-scale electronic structure calculations with density functional theory (DFT) to build an atlas of preferred sites for a range of M(II) and M(III) dopants in the representative III–V InP magic size cluster (MSC). We quantify the thermodynamic favorability of doping, which we identify to be both specific to the sites within the MSC (i.e., interior vs surface) and to the nature of the dopant atom (i.e., smaller Ga(III) vs larger Y(III) or Sc(III)). For isovalent M(III) doping (i.e., Y, Sc, or Ga), we observe stronger sensitivity of the predicted energetics to the ligand binding mode on the surface than to the dopant type. For M(II) (i.e., Zn or Cd) dopants, we show that the binding mode of ligand removed to balance charge during the doping reaction is critical. We observe limited cooperativity with dopants up to moderate concentrations, indicating that rapid single-dopant estimations of favorability from DFT can efficiently guide rational design.