Harder, better, faster, stronger: large-scale QM and QM/MM for predictive modeling in enzymes and proteins
Abstract
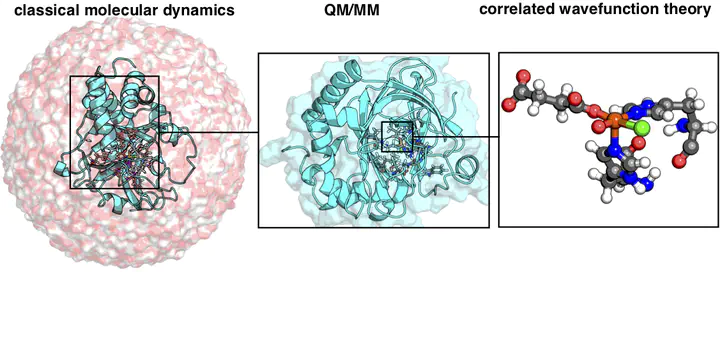
Computational prediction of enzyme mechanism and protein function requires accurate physics-based models and suitable sampling. We discuss recent advances in large-scale quantum mechanical (QM) modeling of biochemical systems that have reduced the cost of high-accuracy models. Trade-offs between sampling and accuracy have motivated modeling with molecular mechanics (MM) in a multi-scale QM/MM or iterative approach. Limitations to both conventional density functional theory (DFT) and classical MM force fields remain for describing non-covalent interactions in comparison to experiment or wavefunction theory. Because predictions of enzyme action (i.e., electrostatics), free energy barriers, and mechanisms are sensitive to the protocol and embedding method in QM/MM, convergence tests and systematic methods for quantifying QM-level interactions are a needed, active area of development.