Quantifying the Long-Range Coupling of Electronic Properties in Proteins with Ab Initio Molecular Dynamics
Abstract
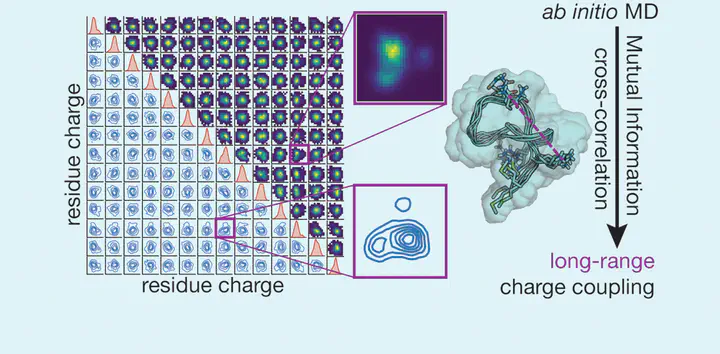
A delicate interplay of covalent and noncovalent interactions gives proteins their unique ability to flexibly play numerous roles in cellular processes. This interplay is inherently quantum mechanical and highly dynamic in nature. To directly interrogate the evolving nature of the electronic structure of proteins, we carry out 100-ps-scale ab initio molecular dynamics simulations of three representative small proteins with range-separated hybrid density functional theory. We quantify the nature and length-scale of the coupling of residue-specific charge probability distributions in these proteins. While some nonpolar residues exhibit expectedly narrow charge distributions, most polar and charged residues exhibit broad, multimodal distributions. Even for nonpolar residues, we observe sequence-specific deviations corresponding to charge accumulation or depletion that would be challenging to capture in a fixed charge force field. We quantify the effect of residue–residue interactions on charge distributions first with linear cross-correlations. We then show how additional insight can be gained from evaluating the mutual information of charge distributions. We show that a significant number of residues couple most strongly with residues that are distant in both sequence and space over a range of secondary structures including α-helical, β-sheet, disulfide bridging, and lasso motifs. The mutual information analysis is necessary to capture coupling between some polar and charged residues. These analyses are expected to be broadly useful in understanding the mechanisms of long-range charge transfer in proteins and for determining what interactions require a quantum mechanical description for predictive simulation of enzyme mechanism and protein function.