Discovering Molecular Coordination Environment Trends for Selective Ion Binding to Molecular Complexes Using Machine Learning
Abstract
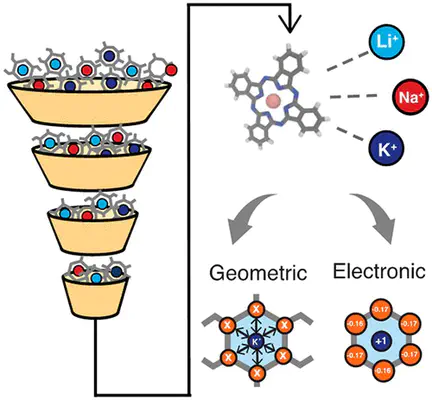
The design of ion-selective materials with improved separation efficacy and efficiency is paramount, as current technologies fail to meet real-world deployment challenges. Selectivity in these materials can be informed by local ion binding in confined membrane ion channels. In this study, we utilize a data-driven approach to investigate design features in small molecular complexes coordinating ions as simplified models of ion channels. We curate a data set of 563 alkali metal coordinating molecular complexes (i.e., with Li+, Na+, or K+) from the Cambridge Structural Database and calculate differential ion binding energies using density functional theory. Using this information, we probe when and why structures favor exchange with alternate ions. Our analysis reveals that energetic preferences are related to ion size but are largely due to chemical interactions rather than structural reorganization. We identify unique trends in the selectivity for Li+ over other alkali ions, including the presence of N coordination atoms, planar coordination geometry, and small coordinating ring sizes. We use machine learning models to identify the key contributions of both geometric and electronic features in predicting selective ion binding. These physical insights offer preliminary guidance into the design of optimal membranes for ion selectivity.