Electronic Structure Origins of Surface-Dependent Growth in III–V Quantum Dots
Abstract
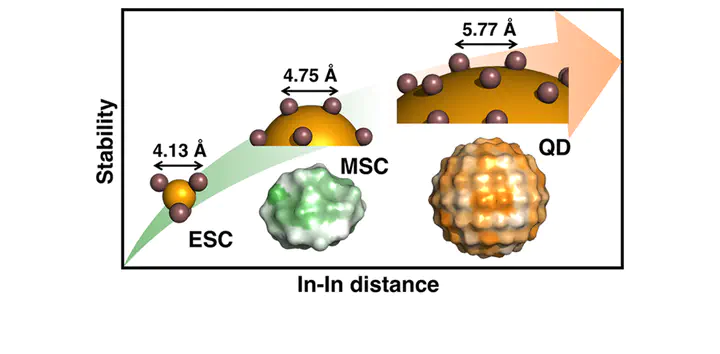
Indium phosphide quantum dots (QDs) have emerged as a promising candidate to replace more toxic II–VI CdSe QDs, but production of high-quality III–V InP QDs with targeted properties requires a better understanding of their growth. We develop a first-principles-derived model that unifies InP QD formation from isolated precursor and early stage cluster reactions to 1.3 nm magic sized clusters and rationalize experimentally observed properties of full sized >3 nm QDs. Our first-principles study on realistic QD models reveals large surface-dependent reactivity for all elementary growth process steps, including In-ligand bond cleavage and P precursor addition. These thermodynamic trends correlate well to kinetic properties at all stages of growth, indicating the presence of labile and stable spots on cluster and QD surfaces. Correlation of electronic or geometric properties to energetics identifies surprising sources for these variations: short In···In separation on the surface produces the most reactive sites, at odds with conventional understanding of strain (i.e., separation) in bulk metallic surfaces increasing reactivity and models for ionic II–VI QD growth. These differences are rationalized by the covalent, directional nature of bonding in III–V QDs and explained by bond order metrics derived directly from the In–O bond density. The unique constraints of carboxylate and P precursor bonding to In atoms rationalize why all sizes of InP clusters and QDs are In-rich but become less so as QDs mature. These observations support the development of alternate growth recipes that take into account strong surface-dependence of kinetics as well as the shapes of both In and P precursors to control both kinetics and surface morphology in III–V QDs.