Abstract
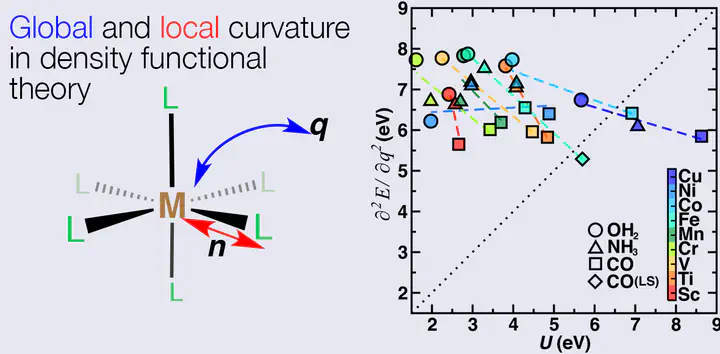
Piecewise linearity of the energy with respect to fractional electron removal or addition is a requirement of an electronic structure method that necessitates the presence of a derivative discontinuity at integer electron occupation. Semi-local exchange-correlation (xc) approximations within density functional theory (DFT) fail to reproduce this behavior, giving rise to deviations from linearity with a convex global curvature that is evidence of many-electron, self-interaction error and electron delocalization. Popular functional tuning strategies focus on reproducing piecewise linearity, especially to improve predictions of optical properties. In a divergent approach, Hubbard U-augmented DFT (i.e., DFT+U) treats self-interaction errors by reducing the local curvature of the energy with respect to electron removal or addition from one localized subshell to the surrounding system. Although it has been suggested that DFT+U should simultaneously alleviate global and local curvature in the atomic limit, no detailed study on real systems has been carried out to probe the validity of this statement. In this work, we show when DFT+U should minimize deviations from linearity and demonstrate that a “+U” correction will never worsen the deviation from linearity of the underlying xc approximation. However, we explain varying degrees of efficiency of the approach over 27 octahedral transition metal complexes with respect to transition metal (Sc–Cu) and ligand strength (CO, NH3, and H2O) and investigate select pathological cases where the delocalization error is invisible to DFT+U within an atomic projection framework. Finally, we demonstrate that the global and local curvatures represent different quantities that show opposing behavior with increasing ligand field strength, and we identify where these two may still coincide.