Where Does the Density Localize in the Solid State? Divergent Behavior for Hybrids and DFT+U
Abstract
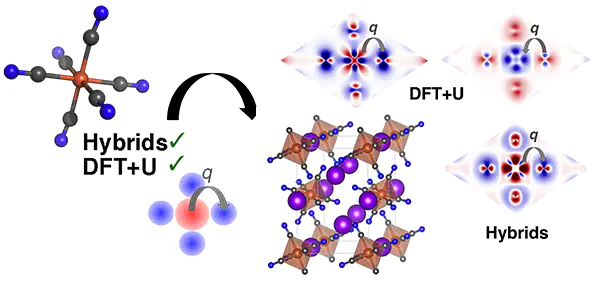
Approximate density functional theory (DFT) is widely used in chemistry and physics, despite delocalization errors that affect energetic and density properties. DFT+U (i.e., semilocal DFT augmented with a Hubbard U correction) and global hybrid functionals are two commonly employed practical methods to address delocalization error. Recent work demonstrated that in transition-metal complexes both methods localize density away from the metal and onto surrounding ligands, regardless of metal or ligand identity. In this work, we compare density localization trends with DFT+U and global hybrids on a diverse set of 34 transition-metal-containing solids with varying magnetic state, electron configuration and valence shell, and coordinating-atom orbital diffuseness (i.e., O, S, Se). We also study open-framework solids in which the metal is coordinated by molecular ligands, i.e., MCO3, M(OH)2, M(NCNH)2, K3M(CN)6 (M = V–Ni). As in transition-metal complexes, incorporation of Hartree–Fock exchange consistently localizes density away from the metal, but DFT+U exhibits diverging behavior, localizing density (i) onto the metal in low-spin and late transition metals and (ii) away from the metal in other cases in agreement with hybrids. To isolate the effect of the crystal environment, we extract molecular analogues from open-framework transition-metal solids and observe consistent localization of the density away from the metal in all cases with both DFT+U and hybrid exchange. These observations highlight the limited applicability of trends established for functional tuning on transition-metal complexes even to equivalent coordination environments in the solid state.