Computational Discovery of Stable Metal-Organic Frameworks for Methane-to-Methanol Catalysis
Abstract
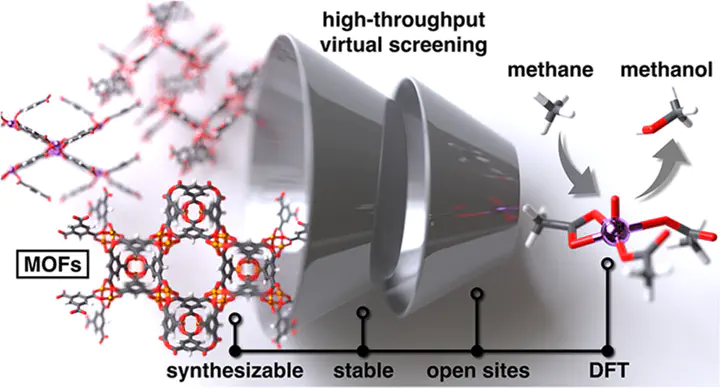
The challenge of direct partial oxidation of methane to methanol has motivated the targeted search of metal–organic frameworks (MOFs) as a promising class of materials for this transformation because of their site-isolated metals with tunable ligand environments. Thousands of MOFs have been synthesized, yet relatively few have been screened for their promise in methane conversion. We developed a high-throughput virtual screening workflow that identifies MOFs from a diverse space of experimental MOFs that have not been studied for catalysis, yet are thermally stable, synthesizable, and have promising unsaturated metal sites for C–H activation via a terminal metal-oxo species. We carried out density functional theory calculations of the radical rebound mechanism for methane-to-methanol conversion on models of the secondary building units (SBUs) from 87 selected MOFs. While we showed that oxo formation favorability decreases with increasing 3d filling, consistent with prior work, previously observed scaling relations between oxo formation and hydrogen atom transfer (HAT) are disrupted by the greater diversity in our MOF set. Accordingly, we focused on Mn MOFs, which favor oxo intermediates without disfavoring HAT or leading to high methanol release energies─a key feature for methane hydroxylation activity. We identified three Mn MOFs comprising unsaturated Mn centers bound to weak-field carboxylate ligands in planar or bent geometries with promising methane-to-methanol kinetics and thermodynamics. The energetic spans of these MOFs are indicative of promising turnover frequencies for methane to methanol that warrant further experimental catalytic studies.