Towards quantifying the role of exact exchange in predictions of transition metal complex properties
Abstract
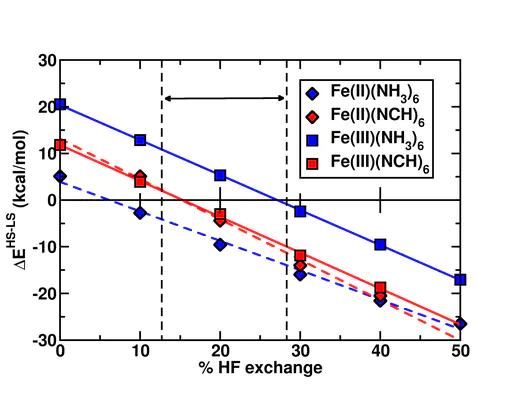
We estimate the prediction sensitivity with respect to Hartree-Fock exchange in approximate density functionals for representative Fe(II) and Fe(III) octahedral complexes. Based on the observation that the range of parameters spanned by the most widely employed functionals is relatively narrow, we compute electronic structure property and spin-state orderings across a relatively broad range of Hartree-Fock exchange (0%-50%) ratios. For the entire range considered, we consistently observe linear relationships between spin-state ordering that differ only based on the element of the direct ligand and thus may be broadly employed as measures of functional sensitivity in predictions of organometallic compounds. The role Hartree-Fock exchange in hybrid functionals is often assumed to play is to correct self-interaction error-driven electron delocalization (e.g., from transition metal centers to neighboring ligands). Surprisingly, we instead observe that increasing Hartree-Fock exchange reduces charge on iron centers, corresponding to effective delocalization of charge to ligands, thus challenging notions of the role of Hartree-Fock exchange in shifting predictions of spin-state ordering.