Density functional theory for modelling large molecular adsorbate–surface interactions: a mini-review and worked example
Abstract
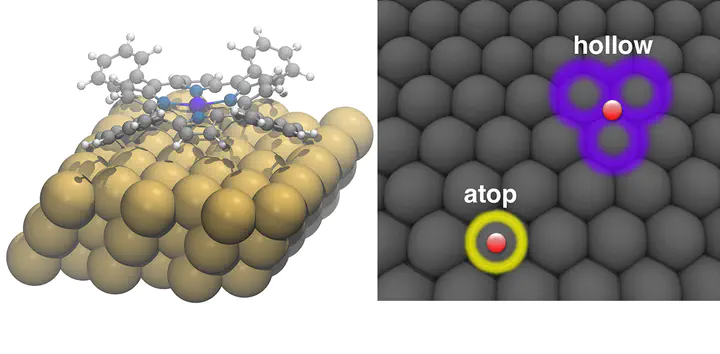
First-principles simulation has played an ever-increasing role in the discovery and interpretation of the chemical properties of surface–adsorbate interactions. Nevertheless, key challenges remain for the computational chemist wishing to study surface chemistry: modelling the full extent of experimental conditions, managing computational cost, minimising human effort in simulation set-up and maximising accuracy. This article introduces new tools for streamlining surface chemistry simulation set-up and reviews some of the challenges in first-principles, density functional theory (DFT) simulation of surface phenomena. Furthermore, we provide a worked example of Co tetraphenylporphyrin on Au(1 1 1) in which we analyse electronic and energetic properties with semi-local DFT and compare to predictions made from hybrid functional and the so-called DFT+U correction. Through both review and the worked example, we aim to provide a pedagogical introduction to the challenges and the insight that first-principles simulation can provide in surface chemistry.