Mechanistic Insights Into Substrate Positioning Across Non-heme Fe(II)/Alpha-Ketoglutarate-Dependent Halogenases and Hydroxylases
Abstract
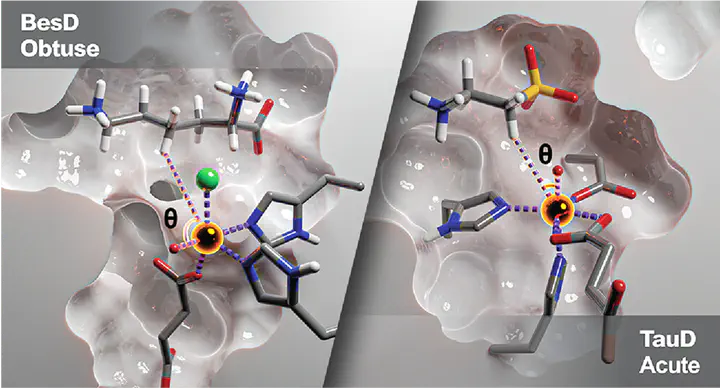
Non-heme iron halogenases and hydroxylases activate inert C-H bonds to selectively catalyze the functionalization of a diversity of biological products under physiological conditions. To better understand the differences in substrate positioning key to their divergent reactivities, we compiled available crystallographic and spectroscopic data, which revealed that hydroxylases prefer an acute oxo-Fe-H target angle, while halogenases prefer a more obtuse angle. With molecular dynamics simulations guided by this experimental information, we simulated the representative hydroxylases TauD and VioC and the halogenases BesD and WelO5 with both acute and obtuse harmonic restraints. We identified key native interactions that maintain the angle of approach in the respective enzymes, such as Asp94 in TauD and His127 in BesD. Moreover, our simulations reveal that the protein environment in halogenases prevents the sampling of acute angles observed in hydrogenases and vice versa. To validate these classical observations, we optimized the structure with large-scale quantum mechanical (QM) simulations and confirmed QM-derived substrate-enzyme hydrogen bond strengths were higher in the native configurations. To understand the effect of positioning on reactivity, we confirmed that reaction barriers for the rate-limiting hydrogen atom transfer reaction was slightly lower from an acute angle regardless of the enzyme-substrate complex. Analysis of the halogenase reaction coordinates reveals the formation of hydrogen bonding networks between the Fe(II)-hydroxyl, monodentate succinate, and a member of the second coordination sphere that may inhibit the hydroxyl rebound.