Harnessing Organic Ligand Libraries for First-Principles Inorganic Discovery: Indium Phosphide Quantum Dot Precursor Design Strategies
Abstract
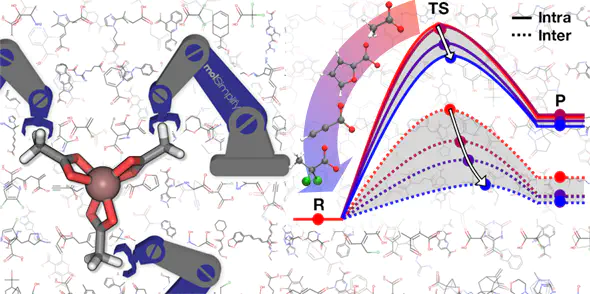
Indium phosphide quantum dots (QDs) represent promising replacements for more toxic QDs, but InP QD production lags behind other QD materials due to limited understanding of how to tune InP QD growth. We carry out a first-principles, computational screen of the tuning of In carboxylate precursor chemistry to alter the kinetics of elementary steps in InP QD growth. We employ a large database normally used for discovery of therapeutic drug-like molecules to discover design rules for these inorganic complexes while maintaining realism (i.e., stable, synthetically accessible substituents) and providing diversity in a 210-molecule test set. We show the In–O bond cleavage energy, which is tuned through ligand functionalization, to be a useful proxy for In–P bond formation energetics in InP QD synthesis. Energy decomposition analysis on a 32-molecule subset reveals that lower activation energies correlate to later transition states, due to stabilization from greater In–P bond formation and more favorable reaction energetics. Our simulations suggest that altering ligand nucleophilicity tunes the reaction barrier over a 10 kcal/mol range, providing the conjugate acid’s pKa as an experimental handle to lead to better control of growth conditions and to improve synthesized InP QD quality. Importantly, these trends hold regardless of phosphorus precursor chemistries and in the longer chain length ligands typically used in synthesis.