New Strategies for Direct Methane-to-Methanol Conversion from Active Learning Exploration of 16 Million Catalysts
Abstract
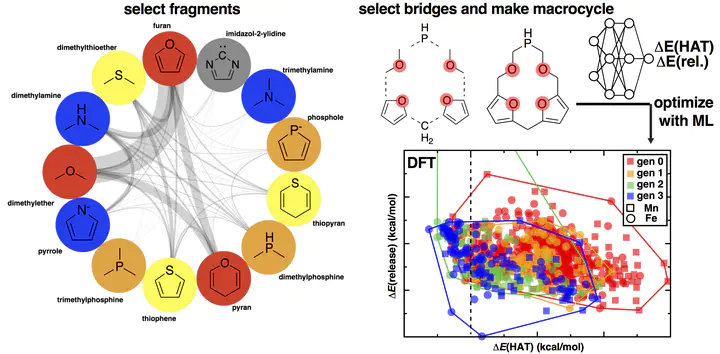
Despite decades of effort, no earth-abundant homogeneous catalysts have been discovered that can selectively oxidize methane to methanol. We exploit active learning to simultaneously optimize methane activation and methanol release calculated with machine learning-accelerated density functional theory in a space of 16 M candidate catalysts including novel macrocycles. By constructing macrocycles from fragments inspired by synthesized compounds, we ensure synthetic realism in our computational search. Our large-scale search reveals that low-spin Fe(II) compounds paired with strong-field (e.g., P or S-coordinating) ligands have among the best energetic tradeoffs between hydrogen atom transfer (HAT) and methanol release. This observation contrasts with prior efforts that have focused on high-spin Fe(II) with weak-field ligands. By decoupling equatorial and axial ligand effects, we determine that negatively charged axial ligands are critical for more rapid release of methanol and that higher-valency metals [i.e., M(III) vs M(II)] are likely to be rate-limited by slow methanol release. With full characterization of barrier heights, we confirm that optimizing for HAT does not lead to large oxo formation barriers. Energetic span analysis reveals designs for an intermediate-spin Mn(II) catalyst and a low-spin Fe(II) catalyst that are predicted to have good turnover frequencies. Our active learning approach to optimize two distinct reaction energies with efficient global optimization is expected to be beneficial for the search of large catalyst spaces where no prior designs have been identified and where linear scaling relationships between reaction energies or barriers may be limited or unknown.