Strategies and Software for Machine Learning Accelerated Discovery in Transition Metal Chemistry
Abstract
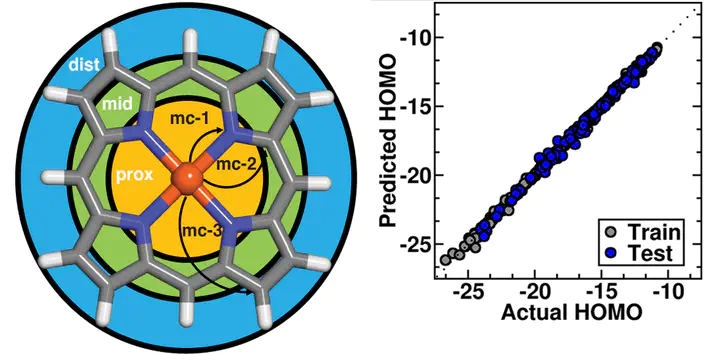
Machine learning the electronic structure of open shell transition metal complexes presents unique challenges, including robust and automated data set generation. Here, we introduce tools that simplify data acquisition from density functional theory (DFT) and validation of trained machine learning models using the molSimplify automatic design (mAD) workflow. We demonstrate this workflow by training and comparing the performance of LASSO, kernel ridge regression (KRR), and artificial neural network (ANN) models using heuristic, topological revised autocorrelation (RAC) descriptors we have recently introduced for machine learning inorganic chemistry. On a series of open shell transition metal complexes, we evaluate set aside test errors of these models for predicting the HOMO level and HOMO–LUMO gap. The best performing models are ANNs, which show 0.15 and 0.25 eV test set mean absolute errors on the HOMO level and HOMO–LUMO gap, respectively. Poor performing KRR models using the full 153-feature RAC set are improved to nearly the same performance as the ANNs when trained on down-selected subsets of 20–30 features. Analysis of the essential descriptors for HOMO level and HOMO–LUMO gap prediction as well as comparison to subsets previously obtained for other properties reveal the paramount importance of nonlocal, steric properties in determining frontier molecular orbital energetics. We demonstrate our model performance on diverse complexes and in the discovery of molecules with target HOMO–LUMO gaps from a large 15,000 molecule design space in minutes rather than days that full DFT evaluation would require.