Are Vanadium Intermediates Suitable Mimics in Non-Heme Iron Enzymes? An Electronic Structure Analysis
Abstract
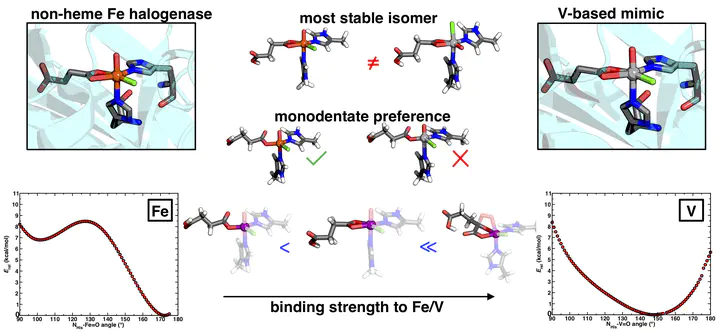
Vanadyl intermediates are frequently used as mimics for the fleeting Fe(IV)═O intermediate in non-heme iron enzymes that catalyze C–H activation. Using density functional theory and correlated wavefunction theory, we investigate the degree to which the electronic structure of vanadium mimics is comparable to that of catalytic iron intermediates. Our calculations reveal crucial structural and energetic differences between vanadyl and ferryl intermediates primarily due to the former having a low-spin ground state and the latter having a high-spin one. This difference in spin state leads to differences in energetics for accessing isomers that confer activity in non-heme hydroxylase and halogenase enzymes. While interconversion between monodentate and bidentate succinate isomers of the key metal-oxo/hydroxo intermediates is energetically favorable for Fe, it is strongly unfavorable in V mimics. Additionally, isomerization of a terminal metal-oxo between an axial and equatorial position is energetically unfavorable for Fe but favorable for V. Analyses of binding strengths of Fe and V intermediates to α-ketoglutarate and succinate co-substrates reveal that both co-substrates bind more strongly to V than to Fe. Given the increasingly frequent use by the biochemistry community of V-based intermediates as mimics, our work highlights the limitations of this approach in studies of non-heme iron enzymes.