How Do Differences in Electronic Structure Affect the Use of Vanadium Intermediates as Mimics in Nonheme Iron Hydroxylases?
Abstract
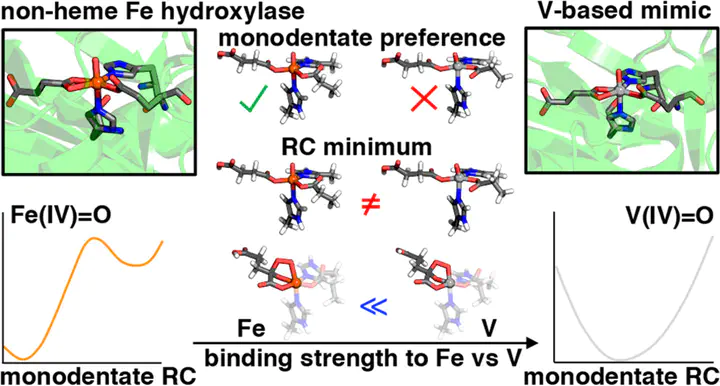
We study active-site models of nonheme iron hydroxylases and their vanadium-based mimics using density functional theory to determine if vanadyl is a faithful structural mimic. We identify crucial structural and energetic differences between ferryl and vanadyl isomers owing to the differences in their ground electronic states, i.e., high spin (HS) for Fe and low spin (LS) for V. For the succinate cofactor bound to the ferryl intermediate, we predict facile interconversion between monodentate and bidentate coordination isomers for ferryl species but difficult rearrangement for vanadyl mimics. We study isomerization of the oxo intermediate between axial and equatorial positions and find the ferryl potential energy surface to be characterized by a large barrier of ca. 10 kcal/mol that is completely absent for the vanadyl mimic. This analysis reveals even starker contrasts between Fe and V in hydroxylases than those observed for this metal substitution in nonheme halogenases. Analysis of the relative bond strengths of coordinating carboxylate ligands for Fe and V reveals that all of the ligands show stronger binding to V than Fe owing to the LS ground state of V in contrast to the HS ground state of Fe, highlighting the limitations of vanadyl mimics of native nonheme iron hydroxylases.