Computational Modeling of Conformer Stability in Benenodin-1, a Thermally Actuated Lasso Peptide Switch
Abstract
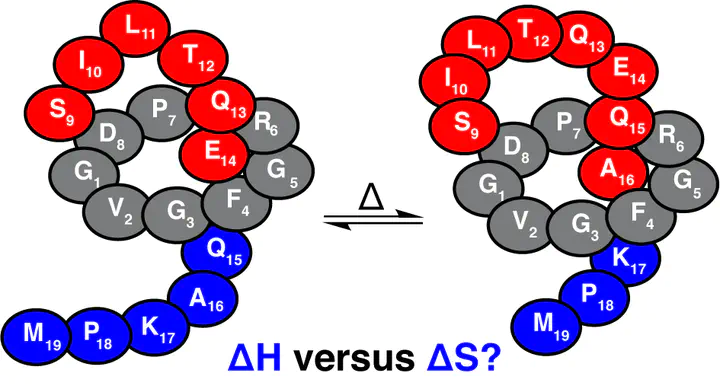
Benenodin-1 is a thermally actuated lasso peptide rotaxane switch with two primary translational isomers that differ in the relative position of the residue Gln15. The conversion from one conformer to the other involves substantial enthalpy–entropy compensation: one conformer is energetically favored and the other is entropically favored. Here, we take a multi-scale quantum mechanical (QM) and classical molecular dynamic (MD) approach to reveal residue-specific sources of these differences in stability. QM reveals that the two benenodin-1 conformers involve distinct hydrogen bonding networks, with the enthalpically favored conformer having more intra-peptide hydrogen bonds between the Gln15 side chain and nearby residues. The evaluation of configurational entropy over the MD-sampled geometries reveals that the entropically favored conformer has enhanced conformational flexibility. By computing the by-residue-sum entropies, we identify the role of Gln15 and neighboring Glu14 in mediating the entropic variation during the switching process. These computational insights help explain the effects of Glu14Ala and Gln15Ala mutations on the conformational population of benenodin-1 observed experimentally.