Modeling the roles of rigidity and dopants in single-atom methane-to-methanol catalysts
Abstract
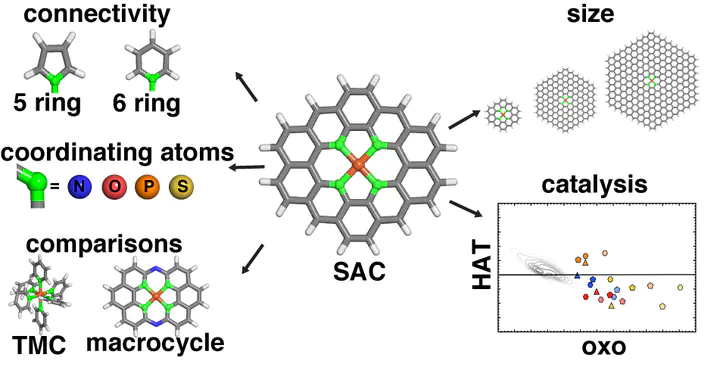
Doped graphitic single-atom catalysts (SACs) with isolated iron sites have similarities to natural enzymes and molecular biomimetics that can convert methane to methanol via a radical rebound mechanism with high-valent Fe(IV) O intermediates. To understand the relationship of SACs to these homogeneous analogues, we use range-separated hybrid density functional theory (DFT) to compare the energetics and structure of the direct metal-coordinating environment in the presence of 2p (i.e., N or O) and 3p (i.e., P or S) dopants and with increasing finite graphene model flake size to mimic differences in local rigidity. While metal–ligand bond lengths in SACs are significantly shorter than those in transition-metal complexes, they remain longer than SAC mimic macrocyclic complexes. In SACs or the macrocyclic complexes, this compressed metal–ligand environment induces metal distortion out of the plane, especially when reactive species are bound to iron. As a result of this modified metal-coordination environment, we observe SACs to simultaneously favor the formation of the metal–oxo while also allowing for methanol release. This reactivity is different from what has been observed for large sets of square planar model homogeneous catalysts. Overall, our calculations recommend broader consideration of dopants (e.g., P or S) and processing conditions that allow for local distortion around the metal site in graphitic SACs.