Abstract
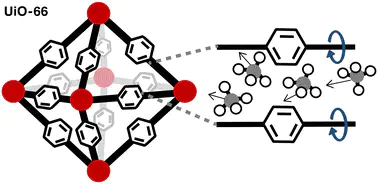
The flexible degrees of freedom in metal–organic frameworks (MOFs) can have significant effects on guest molecule behavior. However, in the majority of studies applying molecular simulations to MOFs, the framework is assumed to be rigid in order to minimize computational cost. Here we assess the significance of this assumption on a representative example of methane uptake and diffusion in UiO-66. We introduce an open-source code to modify MOFs through functionalization and linker rotation and we perform Grand Canonical Monte Carlo and molecular dynamics simulations of methane in each of the functionalized and linker-rotated derivatives of UiO-66. We find that linker rotation moderately influences methane uptake and significantly influences methane diffusion. Our assessment provides ranges of property values that serve as measures of uncertainty of these two properties associated with linker rotation. We further determine that void volume fraction and minimum pore size are the features that govern methane uptake and diffusion, respectively. These findings illustrate the impact of linker rotation on MOFs and provide design principles to guide future investigations.